病例报告│SAMD9基因突变致MIRAGE综合征合并免疫缺陷1例_患儿
本文刊于:中国实用儿科杂志,2026,41(1):84-88
作者:张旖诗,余浪,陶静,赵晓东,安云飞
单位:重庆医科大学附属儿童医院风湿免疫科 国家儿童健康与疾病临床医学研究中心 儿童发育疾病研究教育部重点实验室 感染与免疫罕见病重庆市重点实验室
基金项目:重庆市自然科学基金创新发展联合基金(CSTB2024NSCOLZX0100);重庆医科大学附属儿童医院学科登峰计划临床研究专项揭榜挂帅项目(CHCMU-2024-XKDF-1001)
病例报告│SAMD9基因突变致MIRAGE综合征合并免疫缺陷1例
摘要
回顾2024-12-23重庆医科大学附属儿童医院风湿免疫科收治的1例 SAMD9基因Arg1293Trp突变致MIRAGE综合征患儿资料,分析其临床及实验室检查结果。患儿女,4月龄,以反复肺部感染入院,临床特点有喉软骨发育不良、肾上腺功能不全、胃食管反流和肠道扩张,结合基因测序结果,确诊为MIRAGE综合征。结合已报道的其他5例Arg1293Trp突变患儿免疫学特征分析其死亡结局的原因。造血干细胞移植治疗或许为该患儿晚期惟一的治疗方案。
关键词
SAMD9基因;MIRAGE 综合征;免疫缺陷
MIRAGE综合征最早于2016年由Narumi等[1]定义,用于描述一系列由 SAMD9基因功能获得性杂合突变引起的症状,包括骨髓发育不良、感染、生长受限、肾上腺发育不全、生殖器异常和消化道病变等。MIRAGE综合征并发免疫缺陷的患者通常在生命早期几年内死亡,其免疫异常尚未完全阐明。目前尚无有效治疗方法来降低该病的发生率和病死率,造血干细胞移植的成功率也受到患者先前存在并发症的限制,因此是否需要早期移植仍存在争议[2-3]。本文报道1例由 SAMD9基因p.Arg1293Trp突变导致MIRAGE综合征患儿,重点探讨了该突变引起的免疫缺陷,为该病诊断及治疗提供了更多参考。该研究获得重庆医科大学附属儿童医院伦理委员会批准,伦理委员会批件号:(2021)年论审(研)第(138)号。
1 病历资料
患儿女,4月龄。因“反复肺炎4月余,发热、咳嗽伴痰鸣1 d”于2024-12-23收治于重庆医科大学附属儿童医院风湿免疫科。患儿于入院前4个月(即生后7 d)因面色发绀、炎症指标高,于当地医院治疗,诊断为“新生儿肺炎(重症);新生儿脓毒血症;新生儿胃食管反流;先天性喉软骨发育不良;新生儿甲状腺功能减退症;原发性免疫缺陷病 ?”经住院治疗好转后出院。出院后因反复发热、咳嗽、呛奶、吐奶共计入院4次,1 d前患儿再次发热,最高体温38.9℃,退热处理后能降至正常,有咳嗽,伴痰鸣。为求进一步诊治来笔者所在医院就诊。
个人史:患儿系第5胎第3产,足月剖宫产,出生体重2.42 kg,母亲孕期有甲状腺功能减退症病史,服“优甲乐”治疗。患儿父母及两个哥哥(第3、4胎)均体健,否认家族遗传病史,母亲第1、2胎系社会原因人工流产。生后因喂养困难长期鼻饲空肠管喂养配方奶,仅接种乙肝第1针疫苗。
入院查体:体温36.8 ℃,呼吸52次/min,心率141次/min。 体重4.0 kg,头围37.5 cm,胸围35.0 cm,腹围34.0 cm,身长61.5 cm。发育尚可、营养不良、面色欠佳,头皮胶带状红疹,头枕部可触及一直径3 cm的包块,有波动感。双肺呼吸音稍粗,可闻及痰鸣音及中湿啰音。肛周可见红斑,部分糜烂。外生殖器及神经系统无异常。
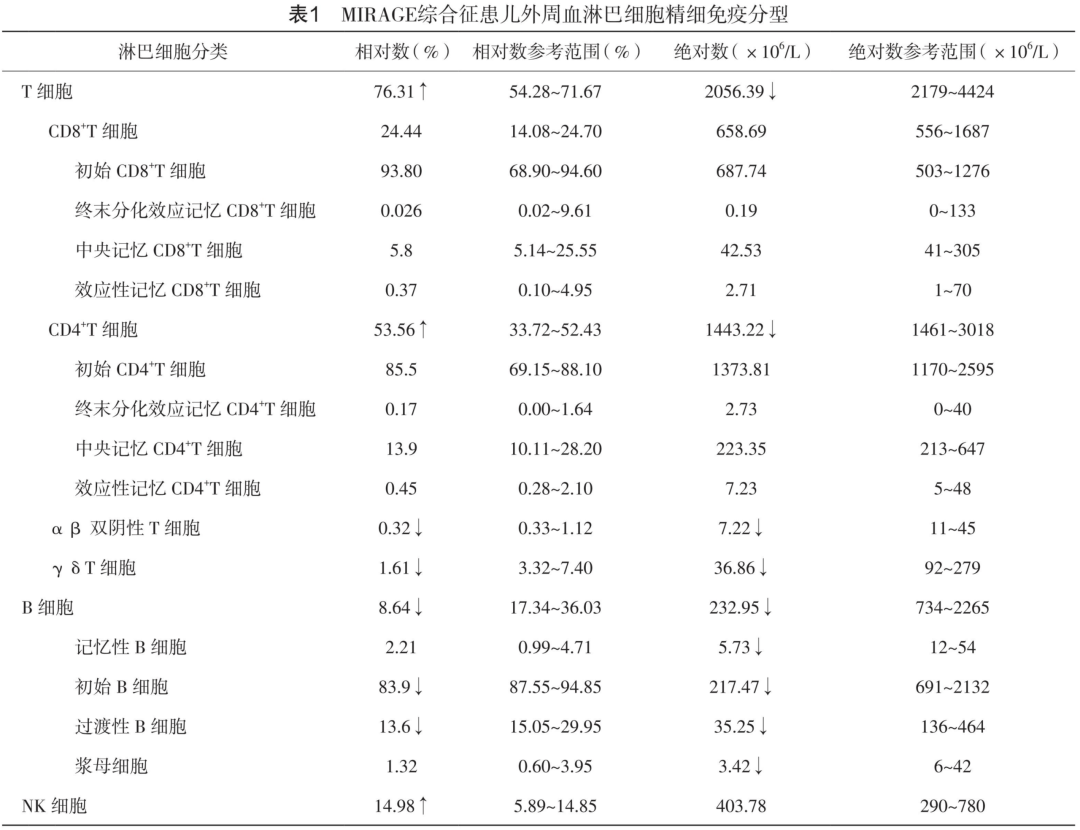
辅助检查:血常规示白细胞计数4.94×109/L,红细胞计数2.75×1012/L↓,血红蛋白95 g/L↓,血小板计数199×109/L,红细胞沉降率31mm/h↑,C反应蛋白5.03 mg/L。 淋巴细胞绝对计数(TBNK):CD3+2056.39×106/L,CD3+76.39%↑,CD3+CD4+53.56%↑,CD4+/CD8+2.19↑,CD19+232.95×106/L↓。 免疫球蛋白示IgG 5.3 g/L↓,IgA 0.115 g/L↓,IgM 0.994 g/L,IgE<5000 U/L,C3 1g/L,C4 0.33 g/L。总甲状腺素38.4 nmol/L↓,类胰岛素样生长因子1<15.0 μg/L↓,促甲状腺素<0.004 mIU/L↓,游离甲状腺素9.27 pmol/L↓,皮质醇381 nmol/L,促肾上腺皮质激素147 ng/L↓。白细胞吞噬功能检测(NBT):正常人无刺激24%,患儿无刺激16%,正常人LPS刺激30%,患儿LPS刺激19%(提示NBT正常);痰培养结果:大肠埃希菌阳性。骨髓细胞学检查提示刺激性骨髓象。常规遗传病染色体检查:46,XX。腹部彩超:胃食管反流,未见幽门肥厚性狭窄。腹部X线摄影:小肠、结肠普遍充气,部分扩张。胸部计算机断层扫描(CT):双肺间质性病变,考虑炎症。鼻咽喉镜:双侧声带外展、内收活动受限,会厌稍卷曲。
基因测序与核型分析:外周血全外显子组测序结果提示该患儿7号染色体 SAMD9基因有一杂合突变:c.3877C>T(p.Arg1293Trp),该突变已被报道为功能获得性突变[2],核型分析提示正常核型(图1a)。
免疫特征分析:外周血精细免疫分型结果提示该患儿各个B亚群细胞绝对值较同年龄幼儿均明显降低(表1),Th2细胞比例减少,Th17比例明显增高(图1b)。T、B细胞增殖实验结果提示该患儿的淋巴细胞增殖功能并没有受到明显的影响(图1c)。
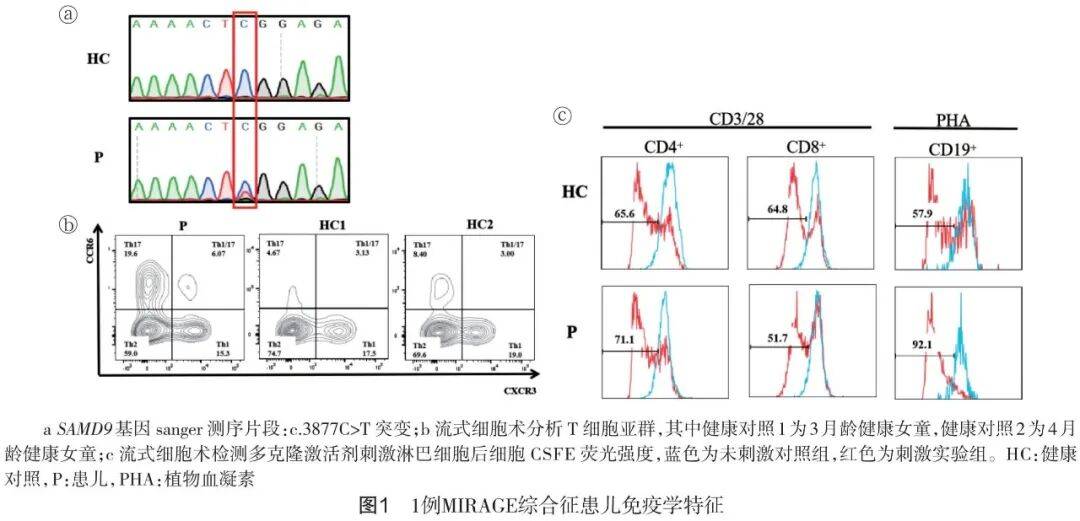
诊断:反复肺炎;原发性免疫缺陷( SAMD9突变);原发性免疫球蛋白缺乏;MIRAGE综合征;先天性喉软骨发育不良;甲状腺功能减退症;房间隔缺损;营养不良;先天性声门关闭不全;足月小样儿(低体重);头皮脓肿;肛周皮炎。
治疗与预后:入院后患儿接受了头皮脓肿切开引流,头孢哌酮钠舒巴坦钠抗感染治疗,静脉注射丙种球蛋白2.5 g,口服氢化可的松、左甲状腺素钠片、谷胱甘肽护肝、奥美拉唑肠溶片和布拉氏酵母菌调节肠道以及雾化和皮肤护理等支持治疗和预防感染等措施,并因喂养困难长期鼻饲喂养。入院14 d后,患儿感染得到控制出院,院外继续使用氢化可的松、左甲状腺素钠片、布拉氏酵母散剂和奥美拉唑肠溶片。鉴于该病目前尚无根治方法,患儿正在等待配型成功后进行造血干细胞移植。
2 讨论
SMAD9基因位于7号染色体的长臂(7q21.2),编码一种生长抑制因子,该因子在调节细胞增殖和抑制肿瘤表型方面发挥作用,该基因的功能获得性突变会导致细胞增殖和蛋白合成被抑制[4-5]。MIRAGE综合征患儿大多存在宫内发育迟缓和早期肾上腺功能不全的症状,对于出现这些症状的患儿,应及时进行基因检测以明确病因。类固醇的早期检测对于预防严重并发症、改善长期预后以及促进患儿的正常生长发育具有关键性作用[6]。同时,肠病的发生可显著影响患儿的生活质量和整体预后[7]。严重感染是MIRAGE综合征患儿最常见的死亡原因,然而大部分患儿并没有表现出明显的血液成分异常,目前导致其感染易感性的机制尚不明确[8]。
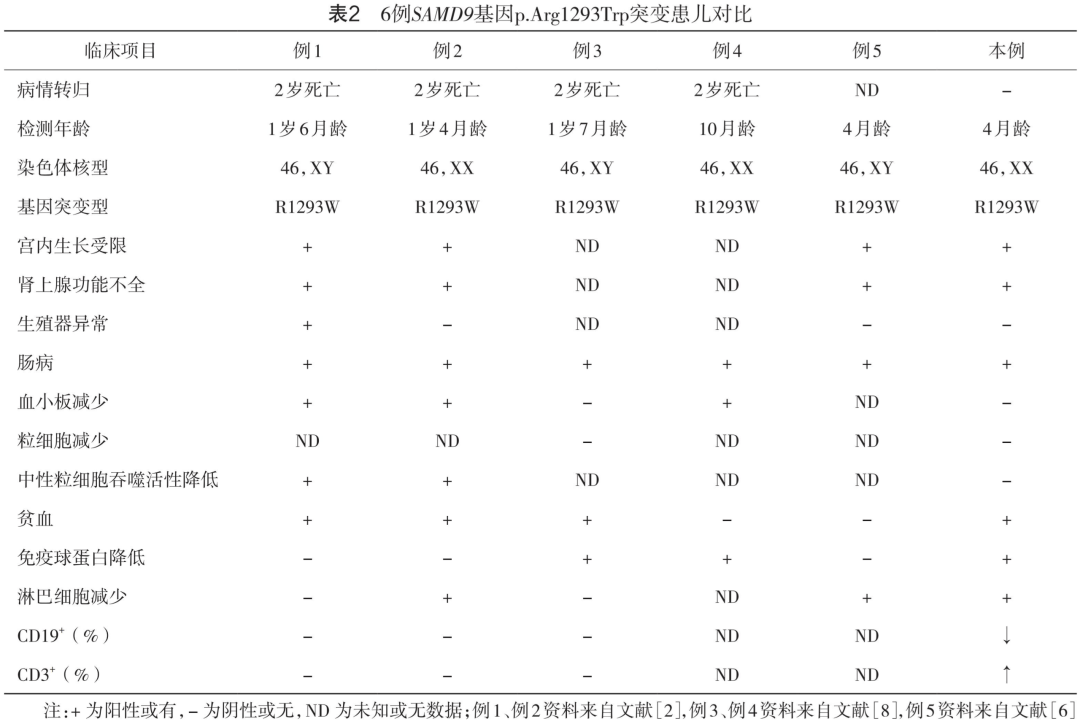
此前已报道了5例 SAMD9基因p.Arg1293Trp突变患儿,6例患儿均出现了肠病、宫内发育迟缓、肾上腺发育不全的表现,其中4例患儿在2岁时因严重感染死亡,1例失去随访[2,8-9]。值得注意的是,其突变位点完全一致,免疫学表型却呈现出异质性。具体来说,3例患者出现了淋巴细胞绝对值降低,3例患儿免疫球蛋白水平下降,而T、B细胞比例异常和显著的Th17细胞增加则是本例患儿特有的表型(表2)。这一差异可能由多种因素导致:一方面这可能与患儿生活环境、种族、检测年龄以及感染情况有关;另一方面,体细胞突变的阶段和比例也可能对结果产生影响。 SAMD9基因上的功能获得性突变可以通过“ 非整倍体适应”机制得到一定程度的中和,即体细胞7号染色体进行性单体化。 这种现象主要发生在 SAMD9/SAMD9L基因,其在一定程度上挽救了功能获得性突变引起的生长抑制,但也同时增加了细胞恶性转化的风险[10-11]。p.Arg1293Trp突变患儿细胞核型检测均正常,这可能与患者年龄尚小,还未出现7号染色体单体细胞累积与扩增有关,是造血干细胞移植的有利条件[12-13]。
MIRAGE综合征患者的预后通常较差,因此适合接受异基因HSCT[14]。目前关于HSCT在合并免疫缺陷的 SAMD9突变患儿中应用的研究较少,此前报道的1例表现为免疫缺陷、中性粒细胞缺乏的 SAMD9p.R824Q突变患儿在26月龄进行HSCT后短期内免疫缺陷改善,但在移植后第440天死于败血症,其在18月龄时,78%的骨髓细胞出现了7号染色体单体[15]。而在其他基因突变型的MIRAGE患儿,鉴于携带p.Arg1293Trp突变的患儿两年存活率为0,且易发生严重感染,本例患儿尚未出现7号染色单体情况,早期进行感染预防干预,并尽早行HSCT 为该患儿最佳治疗方案。治愈性治疗被报道会影响HSCT的结局,因此早期行感染预防干预、营养支持、肠病治疗、激素替代治疗等治疗措施也有益于患儿预后[14]。此外,由于MIRAGE综合征患者7号染色体进行性单体化的特殊性,进行基因监测有助于评估疾病缓解与疾病进展[16-17]。
综上所述,针对宫内发育迟缓和早期肾上腺功能不全的患儿,应警惕MIRAGE综合征,全外显子测序可明确病因。 而对于 SAMD9基因p.Arg1293Trp这类预后极差的基因突变类型的患儿,应尽早行HSCT。
作者贡献声明 张旖诗、余浪收集病历资料、分析免疫学特征、撰写文章;赵晓东、陶静、安云飞:指导诊治,指导论文写作
利益冲突 所有作者声明无利益冲突
(2025-08-22收稿 2025-12-21修回)